- GBS 표준유전체해독과 고밀도 유전지도 작성
-
- Date 2015-10-05
(pseudo-molecule 작성 및 assembly 검정)
그럼에도 거대 지놈을 가진 생물의 표준유전체를 완성하는 일은 많은 비용과 노력이 요구되는 실정입니다. 표준유전체를 조립하는 단계를 살펴보면 Contigs 조립, Scaffolds 조립, 염색체 수준의 Pseudo-molecule 조립 단계를 거치게 됩니다. Contigs와 Scaffolds 조립까지는 NGS 염기서열정보를 In-silico 상의 생물정보분석 기술을 이용하여 진행할 수 있습니다. 그러나 염색체 수준까지 조립하기 위해서는 Genetic map과 Physical map이 필요합니다. BAC clone을 이용한 Physical map 작성은 많은 비용이 들기 때문에, 상대적으로 정교한 고밀도 Genetic map 작성이 요구됩니다.
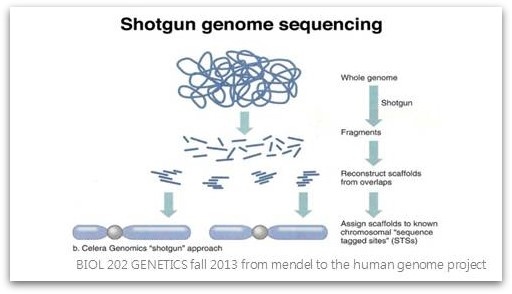
< Shotgun genome sequencing >
◆ 고밀도 유전지도 작성하기
우리는 고추 (3Gb genome size)를 대상으로 resequencing 방식의 고밀도 유전지도작성을 통해 배운 경험을 나누고자 합니다.
먼저 유전지도 작성을 위해서 교배집단을 확보해야합니다. 빈번하게 사용되는 집단은 RIL(Recombination Inbred Line), F2 집단, DH(Double Haploid) 집단입니다. 그러나 과수와 같은 수목은 유전적인 고정에 많은 제약이 있어서 1대 교배실생을 이용하여 pseudo-testcross 기법을 적용합니다.
확보한 집단을 소재로 Resequencing 방식을 이용하는 경우, 유전체 전체의 SNP를 확보할 수 있습니다. 이는 유전지도 작성 시 특정 영역의 결손을 최소화할 수 있는 장점을 가지고 있으나, 너무 많은 수의 SNP가 검출되어 유전지도 작성에 실제로 사용할 정확도 높은 SNP를 선발하는 것이 매우 중요합니다.
◆ Low-depth resequencing 해결
고추 Genome size는 3Gbp 안팎으로 알려져 있습니다. 고추로 Resequencing 기반의 SNP 분석을 수행할 경우, 유전체 전체를 시퀀싱 하기에 비용적인 큰 부담이 있습니다. 이에 우리는 7~9세대 고정된 RIL 집단을 소재로 선정하였고, 비용 부담을 줄이기 위해 모든 계통 고정이 잘 되었을 것이라 가정하고, 1~2x genome coverage로 120계통의 RILs을 시퀀싱 하는 모험을 감행하였습니다.
이 시도는 많은 문제를 유발하는 원인이 있었지만, 이로 인해 생물정보분석 기술의 발전을 이룰 수 있는 계기가 되었습니다. 이 기술은 slide window 방식으로 여러 개의 SNP를 함께 판단함으로써 low depth sequence가 유발할 수 있는 오류를 보정해 정확도 높은 결과를 얻을 수 있었습니다. 이는 고정이 잘된 RIL 계통이었기에 가능했다고 여겨집니다.
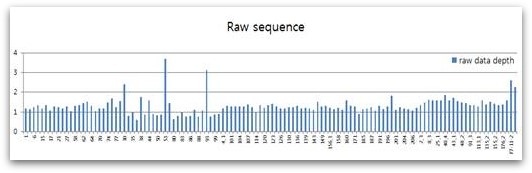
< 120 계통의 sequencing depth >
◆ Missing data를 채우는 것이 관건
분석 결과로 유전체 전체에서 천만 좌(locus) 이상의 SNP를 확보하였습니다. 분석 초기였던 2011년 당시 SNP는 MYSQL DB를 구성하기 힘들 정도로 많았고, 계통 간 결손된 SNP 좌가 너무 많아서 Genetic map을 작성할 수 있는 genotyping matrix 자체도 만들 수 없었습니다. 당시 상당히 많은 고민과 회의가 반복 되었습니다. 그런데 해결할 수 있는 원리는 생각보다 아주 간단했습니다.
그 원리는 ‘recombination이 일어나는 빈도는 수 Mb 이상 떨어져서 일어날 것이다’라는 것이었고 이러한 가정 하에 염기서열을 genotype으로 전환하여 각 계통의 genotype pattern을 인지함으로써 결손 된 SNP 좌를 채워나가는 방식을 반복하여 문제를 해결하였습니다.
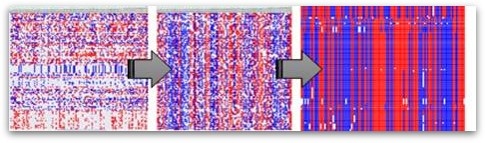
< Genetic map 작성 과정 >
NGS 기술을 이용하면 SNP 선발은 더 이상 문제가 되지 않습니다. 아무리 유사한 계통이어도 수백 혹은 수천 개의 SNP를 찾는 일은 너무 쉬운 일이 되 었습니다. 현재는 너무 많아서 어떤 SNP를 선택할지가 가장 중요한 고민거리입니다. 또한 실험적 genotyping이 아닌 In-silico 상 시퀀스 정보로 genotype을 결정하기에 SNP 정확도가 결과에 미치는 영향이 매우 커졌습니다.
현재 다양한 imputation 기법이 개발되어 공개되어 있는데, 이는 저희가 분석 초기 시도했던 방법과 매우 유사해 보입니다. 단순히 말해서 교배에 의해 자손이 만들어질 때 부모로부터 받은 염색체는 적절한 recombination을 거쳐 각각의 자손 개체가 되므로 그래픽으로 표현하면 recombination map 혹은 bin map의 형태를 띄게 됩니다. 이러한 형태가 나오지 않는다면 SNP 선발 조건을 다시 확인해 봐야 합니다. 이후 결과물을 이용해 linkage group 분석을 반복적으로 수행하여 재현성 여부를 확인하는 방법도 SNP 정확도를 측정하는데 좋은 기준이 될 수 있습니다.
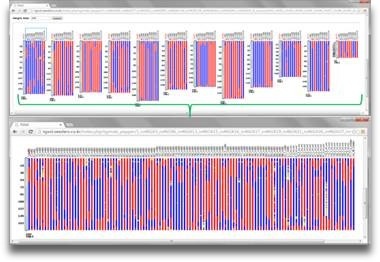
< RIL 집단의 bin map >
◆ 수천 개의 SNP를 이용하여 유전지도 (Genetic map) 작성하기
SNP 수가 많으면 조금 더 정확한 결과를 얻을 수 있는 장점이 있지만, 기존의 프로그램을 이용하는데 많은 제한이 있습니다. 우리는 linkage group을 분석하기 위해 Mapmaker, JoinMap 등의 프로그램을 이용했습니다. 유료 프로그램이지만 편리성 및 분석 속도 면에서 뛰어나기에 JoinMap 4.0을 사용했습니다. 일반적으로 1,000~2,000개의 SNP를 입력하여 분석하는데 큰 문제가 없었으나, 그 이상일 경우 분석 시간이 많이 소요되는 문제가 발생했습니다. 이에 수천 혹은 수만개 SNP를 사용하기 위한 방법으로 linker marker라는 개념을 도입했습니다.
먼저 1,000개 정도의 SNP 마커를 이용해서 여러 개의 map을 만들고, 각각의 map 안에 공통적인 수십 개의 SNP 마커를 넣어서 연결하는 과정을 거치는 방법으로, 실질적으로 프로그램에 입력되는 SNP는 1,000여개지만 수천, 수만개의 SNP 정보를 사용하는 것과 가까운 결과를 얻을 수 있습니다.
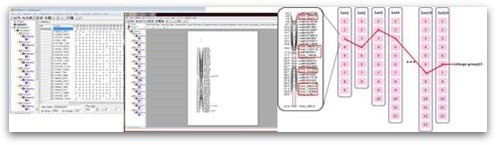
< Genetic map 작성 과정 >
이처럼 많은 어려움을 해결하며 고추 표준유전체해독을 완료하여 최종적인 chromosome 형태를 갖춘 완성도 높은 고추 표준 유전체 (약 3Gb)를 발표하였습니다. 또한 120개의 RIL resequencing 분석을 통한 고밀도 유전지도 성과 Pseudo-molecule 작성을 성공적으로 완료하였습니다. (Kim et al. 2014. Nature Genetics).
최근 GBS 기술 발달이 빠르게 이루어 지고 있습니다. GBS 기술은 기존 Resequencing 방법의 비용적 부담을 줄여주고 다양한 응용연구를 가능하게 하는 신기술로 우리는 GBS 방식으로 유전 지도(Genetic map) 작성을 시도해보았습니다.
이를 통해 배운 경험을 나누고자 합니다. Resequencing 방식과 GBS 방식을 이용한 유전지도 작성 사례를 살펴봄으로 각 방식의 특징과 장단점을 살펴보도록 하겠습니다.